猫冠状病毒药物抑制SARS-CoV-2主要蛋白酶
SARS-CoV-2中的主要蛋白酶M-pro(或3CL-pro)是一个可行的药物靶点,因为它在切割病毒多肽方面起着至关重要的作用。猫感染性腹膜炎是猫的一种致命冠状病毒感染,以前曾成功地用前药GC376治疗,GC376是一种基于二肽的蛋白酶抑制剂。在这里,我们显示前药及其母体GC373是SARS-CoV和SARS-CoV-2的M-pro的有效抑制剂,IC50值在纳摩尔范围内。含有这些抑制剂的SARS-CoV-2Mpro的晶体结构对亲核Cys145具有共价修饰作用。NMR分析表明,抑制是通过半硫缩醛的可逆形成进行的。GC373和GC376在细胞培养中对SARS-CoV-2复制有很强的抑制作用。它们是治疗人类冠状病毒感染的有力候选药物,因为它们已经在动物身上取得了成功。这里的工作为它们在治疗新冠肺炎的人体试验中的使用奠定了框架。
新冠肺炎疫情由于SARS-CoV-2的毒力性质演变成了一场大流行,截至2020年4月底,全球病例达到300多万例,全球感染人数迅速增长1。目前的情况与2002年至2003年爆发的更强但传播效率较低的SARS形成了鲜明对比,当时只有8,000例病例,774人死亡2。目前迫切需要对急性新冠肺炎感染进行抗病毒治疗,特别是在研制出有效的疫苗之前。
冠状病毒是一种RNA病毒,可以劫持宿主的翻译机制来产生病毒蛋白。该病毒编码两个重叠的多聚蛋白:pp1a和pp1ab,其分子量分别为450kD和750kD,分别为450kD和750kD。为了释放病毒复制和转录所需的单个功能蛋白,多聚蛋白需要被切割。病毒编码的蛋白酶包括主要蛋白酶(M Pro),也称为3CL pro和木瓜蛋白酶样蛋白酶(PL Pro)。Mpro主要在保守的Leu Gln|SerAla Gly序列上切割11个位置的多蛋白,这允许病毒复制酶复合物3的组装。
鉴于其在病毒复制中的关键作用,SARS-CoV-2Mpro是新冠肺炎抗病毒治疗的重要药物靶点。冠状病毒Mpro是一种半胱氨酸蛋白酶,有多种不同的抑制剂4。蛋白酶抑制剂如果满足低毒、可溶性和可逆性的要求就是常见的候选药物5。一些蛋白酶已经被确定为分子靶点,并用于开发包括替普那韦在内的治疗HIV 6的新型药物5。然而,除非抑制剂是可逆的,否则用硫醇活性种类抑制半胱氨酸蛋白酶对于人类药物来说往往是站不住脚的。体内不可逆的Michael受体药物,如鲁平曲韦(Rupintrivir),由于生物利用度低而在临床试验中失败。3.大量哺乳动物硫醇发生不受欢迎的不可逆反应来破坏抑制剂。与宿主蛋白硫醇的反应也可能导致急性毒性或免疫反应。在这一点上,硫醇与醛类抑制剂的可逆反应形成半胱氨酸蛋白酶的可逆反应为有效抑制半胱氨酸蛋白酶提供了独特的机会,因为它们可能比其他硫醇7更有效地结合在其目标蛋白的活性部位。此外,水溶性醛亚硫酸氢盐加合物很容易从母醛可逆地生成,并在生理条件下回复8。这些化合物可能是抑制半胱氨酸蛋白酶的理想前药,如下所述。
在早期的研究中,我们开发了针对病毒半胱氨酸蛋白酶7的肽基抑制剂,包括醛,随后在2003年SARS冠状病毒(SARS-CoV)爆发期间用M pro进行了研究。9.肽醛及其亚硫酸氢盐衍生物后来被用来抑制猫冠状病毒FCoV 10的主要蛋白酶。FCoV通常症状轻微,但它可导致猫传染性腹膜炎(FIP),这在猫中通常是致命的。亚硫酸氢盐加合物GC376是一种容易转化为肽醛GC373的前药,耐受性良好,能够阻断猫11的感染。这与包括雪貂和水貂冠状病毒Mpro在内的其他研究一起,证明了这种蛋白酶抑制剂10、12、13的广泛特异性。用同源的中东呼吸综合征(MERS)Mpro解析了GC376的晶体结构,并证明了GC376与MERS催化半胱氨酸的共价相互作用。这些Mpro抑制剂还没有在冠状病毒感染的动物模型中进行测试,也没有在SARS的人类或动物试验中报道。已知许多Mpro抑制剂
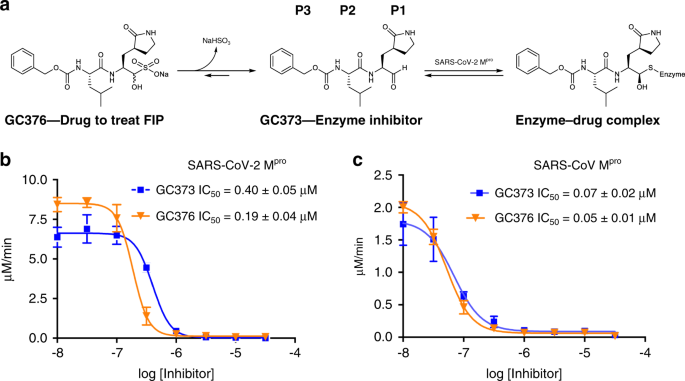
IC50测量表明,GC373和GC376在体外均在纳摩尔浓度下抑制SARS-CoV-Mpro和SARS-CoV-2Mpro(图1b,c)。对SARS-CoV-2M PRO,GC373和GC376的IC50分别为0.4 0 ± 0.0 5 μM和0.19 ± 0.0 4 μM。这与GC376与Mpro在相关病毒中的研究是一致的。对FCoVMpro的IC50为0.070.49 ± 0.0 7 μM,而对雪貂和水貂冠状病毒Mpro的IC50分别为1.33 ± 0.19 μM和1.4 4 ± 0.38 μM。12.对SARS冠状病毒Mpro有增强的IC50,两种化合物均有广泛的抑制作用,GC373和GC376的IC50分别为0.33 ± 0.0 2 μM和0.0 5 ± 0.0 1 μ。与游离醛相比,亚硫酸氢盐加合物GC376对这两种酶的效力略高。我们对GC373和GC376的体外IC50值表明,与其他体外测试的抑制剂相比,Ebselen(IC500.67 μM)16,tidelusib(IC501.55 μM)16,卡莫氟(IC501.82 μM)16,二磺兰(IC509.35 μM)16,紫草素(IC5015.75 μM)16,PX-12(IC5021.39 μM)16.GC373和GC376也比最近报道的酮酰胺抑制剂(先导化合物的IC50为0.67 μM)15.最近报道了一种相关的肽基抑制剂,与我们的化合物具有相似的弹头,但在P3位有吲哚基团,IC50为0.05 ± 0.005 μM18.然而,该化合物尚未被证明对动物有效,GC3也是如此。
为了深入了解抑制机制,用抑制剂GC373M和GC376MPRO晶体结构分别在2.0kPa和1.9kPa被测定(图2和补充表A1),正如预期的那样,前药物转化为药物,导致结构中有相同的配体。(2)为了深入了解抑制机制,我们分别测定了含有抑制剂GC373M和GC376M的PRO晶体结构(图2和补充表A1),结果在结构中产生了相同的配体。ApoSARS-CoV-2M PRO(PDBcode 6WTM)的三维结构与最近解决的结构高度相似,均方根偏差为0.19PDBcode(PDBcode 6Y2E)15,16。SARS-CoV-2M预先结晶为二聚体,由原生质体A(残基1-7)的N指促进,该二聚体适合于原生质体B的口袋。每个原生质体都表现为两叶结构,其中一叶由两个反平行的两个叶组成。 ä(PDB Code 6Y2E)15,16.SARS-CoV-2M预先结晶为二聚体,该二聚体由两个反平行的原始体A(残基1-7)的N指促进。活性位点由位于结构域界面的Cys145-His41二联体组成。受二聚作用19影响的氧阴离子空穴是由主链残基Gly143、Ser144和Cys145形成的。C-末端结构域III参与结构域交换,促进二聚体形成20。6Y7M.PDB的分子置换显示,前体药物GC376(PDB代码6WTJ)和药物GC373(PDB代码6WTK)在催化半胱氨酸的Fo-FC图谱上都有电子密度。在这两个结构中,肽基抑制剂以半硫缩醛的形式共价连接到Cys145上,表明亚硫酸氢基离开了GC376(补充图17.)。与MERS M pro-GC376结构相比,SARS-CoV-2M pro电子密度表明该抑制剂14只形成一个对映体,Gly143、Ser144和Cys145的骨架贡献定义了氧阴离子空穴。氧阴离子空穴通过β-Turn进一步稳定,其中Leu141的羰基与Ser144的主链胺结合,Leu141的主链酰胺与Ser144的羟基相互作用(图3a)。结构域II中的残基建立了疏水相互作用和氢键网络,以支持抑制剂的结合。P2插入由总碱基His41和代表酶S2亚位点的残基Met49和Met165组成的疏水口袋中。(图3b)。底物P1位的谷氨酰胺替代物与His163和Glu166的侧链形成氢键,并与His172形成疏水相互作用。P3中的羰基与Glu166的主链酰胺形成氢键(图3c,d)。与在MERS M pro-GC376Structure 14中观察到的相似,谷氨酰胺替代物的苄环和γ-内酰胺形成堆积的疏水相互作用,使抑制剂稳定在蛋白酶的活性位点。总之,这为抑制剂提供了很强的结合力和较低的IC50。仔细检查SARS-CoV-2MPRO的子站点,可以发现允许未来开发抑制剂的区域(补充图17)。
Apo-SARS-CoV-2M蛋白形成二聚体(6WTM.pdb)。B前药GC376与SARS-CoV-2Mpro孵育后转化为GC373,GC373与C145形成共价键。表面表示揭示了与GC373(6WTJ.pdb)的络合物中的活性中心口袋。一个SARS-CoV-2原生质体与抑制剂GC373结合在Ⅱ区的复合物中的c条带表达与SARS-CoV-2 M pro的活性位点半胱氨酸发生共价相互作用,D GC373与SARS-CoV-2 M pro的活性位点半胱氨酸发生共价相互作用。1σ时的电子密度以灰色网格显示。
GC373与C145形成共价键,氧原子
许多药物最初被设计用来抑制SARS-CoV M PRO3,然而,2002年SARS的爆发受到公共卫生措施的控制,这些化合物从未获得许可。GC376已成功地用于治疗猫的FIP,其分解产物GC373有效地抑制了FCoV的Mpro。我们的研究表明GC376和GC373是SARS-CoV-2Mpro的有效抑制剂,与先前的研究表明GC373和GC376对冠状病毒和钙化病毒的Mpro具有广泛的特异性(补充表2)10,13,14,22。我们的研究表明,GC376和GC373对冠状病毒和钙化病毒的Mpro具有广泛的特异性(补充表2)10,13,14,22。我们的研究表明,GC376和GC373对冠状病毒和钙化病毒的 μ-2Mpro具有广泛的特异性(补充表2)10,13,14,22。显然,这些药物需要迅速推进到新冠肺炎的人体试验中。SARS-CoV-2是新冠肺炎的病因,是一种具有显着变异率23的病毒。此外,在一些患者中,病毒持续了2个月以上,并有一定的再次感染的可能性24。疫苗至关重要,但仍可能需要一年或更长时间,因为这种病毒可能会对疫苗构成挑战。
有许多临床试验测试从原来的适应症改变用途的药物。雷米德韦是一种用于治疗埃博拉病毒25的聚合酶抑制剂,最初用于同情病例,现在美国食品和药物管理局批准用于新冠肺炎26岁患者的紧急使用。另一种用于抑制RNA依赖的RNA聚合酶(包括冠状病毒中的RNA聚合酶)的药物是EIDD2801(一种N4-羟基胞苷三磷酸,被掺入病毒RNA中以促进后代RNA中的错误)。对于新冠肺炎来说,这些直接作用的抗病毒药物(DAA)的例子是至关重要的27。GC373和GC376也都是专为冠状病毒设计的DAA。很可能需要几种非常有效的药物组合来治疗SARS-CoV-2,并防止耐药突变28的放大,就像HIV 29的情况一样。本研究结果表明,GC373和GC376是新冠肺炎应加速进入临床试验的候选抗病毒药物。
补充方法中报告了协议、合成细节、化合物表征和相关的方法学信息。所有化合物的鉴定和纯度(>;95%)由色谱(硅胶或RP-18HPLC)、全归属的1H和13C-NMR谱和高分辨质谱确定。
编码SARS-CoV-2主要蛋白酶Mpro的DNA(Genbank:MN908947.3)由BioBasic公司获得。该基因位于加拿大安大略省),包含融合基因上游的EcoRI酶切位点和下游的HindIII酶切位点。对M pro基因进行密码子优化,使其在大肠杆菌中表达。将该融合基因克隆到pE的EcoRI和HindIII酶切位点上。
.