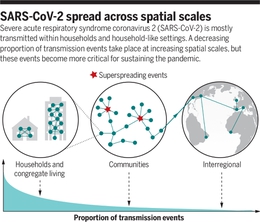
数据分析确定了所有SARS-CoV-2基因组的“母亲”

在分子流行病学领域,世界科学界一直在努力解开SARS-CoV-2早期历史之谜。自2019年12月检测到第一例SARS-CoV-2病毒感染以来,全球已对其数万个基因组进行了测序,显示这种冠状病毒正在以每年25例的速度变异,尽管速度很慢。
但是,尽管付出了巨大努力,到目前为止还没有人确认首例人传播病例,也没有人在新冠肺炎大流行中发现过零患者。找到这样的病例是必要的,以更好地了解病毒可能是如何首先从动物宿主跳到感染人类的,以及SARS-CoV-2是如何随着时间的推移变异并在全球传播的历史。
坦普尔大学(Temple University)基因组与进化医学研究所所长苏迪尔·库马尔(Sudhir Kumar)说,SARS-CoV-2病毒携带的RNA基因组已经感染了全球3500万人。我们需要找到这个共同的祖先,我们称之为祖先基因组。
在没有零号患者的情况下,库马尔和他的坦普尔大学(Temple University)研究团队现在可能已经找到了下一个最好的东西来帮助全世界的分子流行病学检测工作。这项研究的资深作者Sayaka Miura说,我们开始利用从感染者身上获得的冠状病毒基因组的大数据集来重建祖先的基因组。
他们发现,所有SARS-CoV-2基因组的母株及其早期后代毒株随后都发生了突变,并在全球范围内蔓延。库马尔说,我们现在已经重建了祖先基因组,并绘制了最早的突变发生的地点和时间。库马尔是一项预印研究的通讯作者,该研究可以在biRxiv服务器上找到。
通过这样做,他们的工作为SARS-CoV-2的早期变异历史提供了新的见解。例如,他们的研究报告称,SARS-CoV-2刺突蛋白(D416G)的突变,通常与传染性和传播的增加有关,发生在新冠肺炎开始工作几周后的许多其他突变之后。这项研究的资深合著者谢尔盖·庞德(Sergei Pond)表示,它几乎总是与许多其他蛋白质突变一起被发现,因此它在增加传染性方面的作用仍然很难确定。
除了他们对SARS-CoV早期病史的发现外,库马尔的团队还开发了突变指纹,以快速识别感染个人或在全球范围内定居的菌株和亚型。
为了识别先祖基因组,他们使用了突变顺序分析技术,该技术依赖于对突变株的克隆分析,以及SARS-CoV-2基因组中两对突变一起出现的频率。
首先,库马尔的团队筛选了近30,000个SARS-CoV-2病毒完整基因组的数据。SARS-CoV-2是导致新冠肺炎的病毒。他们总共分析了29681个SARS-CoV-2基因组,每个基因组包含至少28000个碱基的序列数据。这些基因组是在2019年12月24日至2020年7月7日期间采集的,代表全球97个国家和地区。
库马尔说,之前分析如此庞大的数据集的许多尝试都没有成功,因为人们专注于构建SARS-CoV-2的进化树。这种冠状病毒进化太慢,需要分析的基因组数量太多,而且基因组的数据质量变化无常。我立刻发现这些冠状病毒基因数据的特性与另一种邪恶疾病--癌症的克隆传播的基因数据有相似之处。
库马尔的团队已经开发和研究了许多用于分析癌症患者肿瘤基因数据的技术。他们对这些技术进行了调整和创新,并建立了一条自动追溯到祖先的突变轨迹。库马尔说,基本上,第一次突变之前的基因组是祖先的基因组。突变追踪方法很漂亮,可以预测SARS-CoV-2主要毒株的系统发育。这是一个很好的例子,说明了大数据与生物信息数据挖掘相结合是如何揭示重要模式的。
库马尔的团队发现了所有SARS-CoV-2基因组(ProCoV2)的祖细胞(母亲)基因组的预测序列。在proCoV2基因组中,他们确定了170个非同义突变(导致蛋白质中氨基酸变化的突变)和958个同义替换,与在蝙蝠中发现的密切相关的冠状病毒RaTG13的基因组相比。虽然从蝙蝠到人类的中间动物尚不清楚,但proCoV2和RaTG13序列的相似性达到了96.12%。
接下来,他们从他们的数据集中识别了49个(SNV),这些SNV的变异频率超过1%。对这些基因进行了进一步检查,以观察它们的突变模式和全球传播情况。
库马尔说,突变之树预示着一棵菌株之树。你也可以先做菌株树,然后预测突变的顺序。然而,这种方式很大程度上受序列质量的影响。当突变率很低时,很难区分低质量导致的错误和真正的突变。我们采取的方法对测序错误的抵抗力要强得多,因为对基因组中位置对的分析提供了更多的信息。
库马尔的研究小组将推断出的proCoV2序列与他们收集的基因组进行比较,发现在核苷酸水平上没有完全匹配的时候,他们知道疫情开始的最初时间是错的。
库马尔说,这个祖先基因组的序列与一些人所说的参考序列不同,参考序列是在中国首次观察到的,并被存入GISAID SARS-CoV-2数据库。
最接近的匹配是在2019年12月24日最早的采样病毒上市12天后采样的基因组。在所有抽样的大陆都发现了多个匹配,直到2020年4月才在欧洲检测到。总体而言,库马尔的团队分析了120个基因组,所有这些基因组都只包含与proCoV2同义的差异。也就是说,它们的所有蛋白在氨基酸序列上都与相应的proCoV2蛋白相同。在这些蛋白质水平的匹配中,大多数(80个基因组)来自中国和其他亚洲国家的冠状病毒样本。
这些时空模式表明,proCoV2已经拥有了感染、传播和在全球人类群体中持续存在所需的全部蛋白质序列。
他们发现,基于proCoV2最早的突变及其位置,proCoV2病毒及其最初的后代出现在中国。此外,他们还证明,在中国首次发现新冠肺炎病例时,存在与原CoV2有多达6个突变差异的毒株群体。据估计,SARS-CoV-2每年变异25次,这意味着该病毒肯定在2019年12月病例发生前几周就已经感染了人们。
由于在参考基因组中发现的突变之前有强有力的证据表明存在许多突变,库马尔的团队不得不提出一个新的突变特征命名法来对SARS-CoV-2进行分类,并通过引入一系列希腊字母符号来代表每一个突变来解释这些突变。
例如,他们发现μ和αSARS-CoV-2基因组变异的出现早于新冠肺炎的第一次报告。这有力地暗示了SARS-CoV-2祖先群体中存在一定的序列多样性。2019年12月从中国采集的所有17个基因组,包括指定的SARS-CoV-2参考基因组,都携带所有三个μ和三个α变体。有趣的是,这六个含有μ变异体但不含α变异体的基因组于2020年1月在中国和美国进行了抽样。因此,最早采样的基因组(包括指定的参考)不是祖先菌株。
它还预测,祖先基因组的后代在新冠肺炎的早期阶段就在世界各地传播。它从一开始就准备好感染了。
谢尔盖·庞德(Sergei Pond)说,祖先拥有传播所需的一切能力。虽然蝙蝠对冠状病毒有很强的选择性,但几乎没有证据表明蝙蝠和人类在血统上存在选择。
此外,他们发现了令人困惑的证据,即D416G尖峰蛋白突变总是伴随着另一种突变。
库马尔说,许多人对尖峰蛋白的突变感兴趣,因为它的功能特性。但我们观察到的是,除了尖峰蛋白之外,基因组中还有几个额外的变化,这些变化总是伴随着尖峰蛋白(D416G)的变化而出现的。我们称之为一组贝塔突变,尖峰突变就是其中之一。无论我们认为尖峰突变在做什么,最好不要忘记也可能涉及其他突变。或者,这些突变可能只是搭便车而已,我们还不能断定。
同样有趣的是,含有尖峰蛋白突变的基因组经历了许多其他突变。我们所说的epsilon突变(有3种)发生在尖峰突变的背景下,它们改变了一种非常重要的蛋白质--核衣壳(N)蛋白中的精氨酸残基。Epsilon突变在欧洲很普遍,它们总是伴随着尖峰蛋白突变而被发现。因此,epsilon突变在欧洲和亚洲开始了占主导地位的分支。
他们总共确定了疫情开始后出现的七个主要进化谱系,其中一些是在中国祖先谱系起源之后出现在欧洲和北美的。
库马尔说,亚洲病毒株导致了整个疫情。但随着时间的推移,可能发生在中国境外(最先在中东和欧洲发现)的是含有epsilon突变的亚型毒株,它对亚洲的感染要严重得多。
他们基于突变的分析还证实,北美冠状病毒与欧洲和亚洲流行的冠状病毒具有非常不同的基因组特征。
库马尔说,这是一个动态的过程。显然,新突变的出现描绘了截然不同的传播图景,我们发现这三种突变发生在尖峰蛋白变化之后。我们需要找出这些突变的任何功能特性是否加速了大流行。
庞德说,目前已有超过10万个SARS-CoV-2基因组被测序。库马尔说,这种方法的强大之处在于,你拥有的数据越多,你就越容易分辨出单个突变和突变对的精确频率。这些产生的变异,单核苷酸变异,或SNV,它们的频率和历史可以通过更多的数据很好地描述出来。因此,我们的分析为SARS-CoV-2的系统发育提供了一个可靠的根源。
随着新基因组的报道,他们的结果正在自动在线更新(现在超过50,000个样本,可以在http://igem.temple.edu/COVID-19).上找到
库马尔说,这些发现和我们对SARS-CoV-2毒株的直观变异指纹克服了艰巨的挑战,可以追溯新冠肺炎是如何、何时以及为什么出现并传播的,这是通过科学、技术、公共政策和医学的努力创造出战胜这种流行病的药物的先决条件。更多信息:Sudhir Kumar等人。新冠肺炎大流行中SARS-CoV-2及其主要分支的进化图景,(2020年)。地址:10.1101/2020.09.24.311845。
引用:数据分析确定了2020年11月5日从https://medicalxpress.com/news/2020-11-analysis-mother-sars-cov-genomes.html检索到的所有SARS-CoV-2基因组中的母亲。
本文档受版权保护。除为私人学习或研究的目的而进行的任何公平交易外,未经书面许可不得复制任何部分。提供的内容仅供参考。